
C1: Jablonski diagram
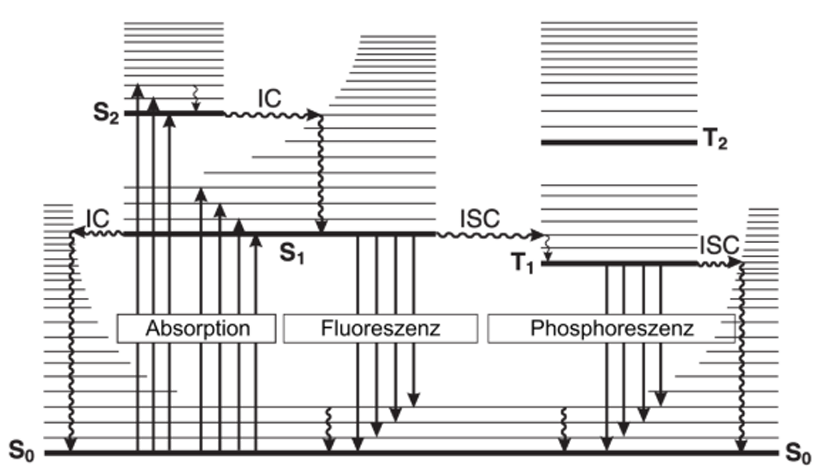
Various relaxation processes can take place from the excited state. The possible transitions are illustrated using a simplified energy level diagram, the so-called Jablonski diagram (see Figure 1).[1]
Excitation
Most systems are usually in the singlet ground state S0. By absorbing a photon, the system is excited into an energetically higher electronic state Sn. The spin multiplicity is maintained according to the selection rules and a spin-allowed S0 → Sn (n > 0) transition occurs. After excitation, the system also occupies an oscillation level ν of the Sn state. However, vibrational transitions are not subject to strict selection rules when switching between two different electronic states. A statement about the probability with which a vibronic state is occupied can be made using the Franck-Condon principle.
Relaxation transitions
Starting from the excited state, there are several processes that can occur to relax to the singlet ground state. In general, these can be divided into radiative and non-radiative processes. In most cases, these processes compete after reaching the vibrational ground state of the S1 state. According to Kasha's rule, photons are emitted from the lowest excited state of a multiplicity (valid for both singlet and triplet states).[3] However, this rule is empirical and does not apply to all systems.
Internal conversion - IC
The S1 state is achieved through radiation-free vibrational relaxation and internal conversion. At a speed of 10−12 - 10−10 s, the vibrational relaxation is very fast and therefore unrivalled. The internal conversion then occurs from the lowest vibronic state. This is a transition between two electronic states with the same spin multiplicity. The electronic energy of the initial state is converted into the vibronic energy of the final state, whereby the system is then in a higher vibronic state. This transition is radiationless as the vibronic levels of the initial and final states are isoenergetic. This is followed by vibrational relaxation. This process continues until the S1 state is reached at its lowest vibrational level or until the molecule is thermalised. The Sn+1→ Sn transitions are more efficient than the S1 → S0 transition due to the smaller energy differences between these states. The internal conversion takes place in a speed range of 10−11 - 10−9 s, but the large energy gap between the S0 and S1 state leads to a slowdown due to the lower efficiency. This enables competition with the processes of intersystem crossing (ISC) with a speed range of 10−10 - 10−8 s and fluorescence with a lifetime of 10−10 - 10−7 s.
Fluorescence
The radiative deactivation process into the singlet ground state S0, while maintaining multiplicity, is fluorescence. The transition takes place under the emission of a photon, which is usually a S1 → S0 transition. After the transition, as with excitation, different vibronic states are occupied according to the Franck-Condon principle. Exceptions such as fluorescence from the S2 state are shown by the example of azulene.[4] There are also substances that emit from both the S1 and the S2 state with a balanced ratio. Such 'balanced non-Kasha dual emission' can be observed, for example, in molecules of the triphenylmethane dye group.[5,6]
Intersystem crossing – ISC
A process that is similar to internal conversion is intercombination, which is also radiationless. This is also a transition between two isoenergetic vibration levels, but they belong to electronic states with different multiplicities. The transition from a singlet to a triplet state is formally spin-prohibited, but can occur with large spin-orbit coupling. Molecules containing atoms with high atomic mass such as Br, Pt, Ir, Au can exhibit strong spin-orbit coupling, which increases the efficiency of intercombination and thus the quantum yield and intensity of phosphorescence.[7,8]
Phosphorescence
After a S1 → Tn transition, radiationless vibrational relaxation and internal conversion to the T1 state at the lowest vibrational level occurs according to Kasha's rule. This can be followed by a T1 → S0 transition with the emission of a photon. This emission is phosphorescence. The occupation of the oscillation levels in the S0 state is analogous to that in fluorescence. The wavelength of the emitted radiation is usually lower in phosphorescence compared to fluorescence, as the T1 state is lower in energy than the S1 state. Since the T1 → S0 transition is spin-forbidden, the radiation rate constant is very low. This means that the lifetime of the triplet state can be much longer than that of the singlet state. They are in the range of 10−10 - 10−7 s for the singlet state. However, the lifetime of the triplet state can be in the range of 10−6– 10 s.
Other relaxation pathways
In addition to the common processes, there are other rarer processes such as triplet-triplet transitions or delayed fluorescence. After an excited system occupies the T1 state, a spin allowed transition to an energetically higher triplet state Tn can occur through the absorption of photons. The T1 states are usually metastable states with a sufficient population.[9] Such triplet-triplet transitions can occur in polyatomic molecules such as benzene or naphthalene.[10]
Thermally activated delayed fluorescence (TADF) occurs when the energy difference between the T1 and S1 state is small and the lifetime of the T1 state is sufficiently long. A T1 → S1 transition occurs with subsequent fluorescence. This process is thermally activated and is favoured at higher temperatures. The delayed fluorescence behaves analogue to the ordinary fluorescence, the only difference is the longer decay time constant, which is caused by the additional S1 → T1 and T1 → S1 transitions. This case was observed for the first time with eosin.[11,12]
Life times
Table 1: Characteristic life times of the processes shown in Figure 1. [13]

References
[1] A. Jablonski, Nature 1933, 131, 839–840.
[2] D. Graf, Master Thesis, Karlsruher Institut für technologie (KIT), 2023.
[3] M. Kasha, Discuss. Faraday Soc. 1950, 9, 14–19.
[4] M. Beer, H. C. Longuet-Higgins, J. Chem. Phys. 1955, 23, 1390–1391.
[5] A. Janowski, J. Rezeszotarska, J. Lumin. 1980, 21, 409–416.
[6] S. K. Behera, S. Y. Park, J. Gierschner, Angew. Chem. Int. Ed. 2021, 60, 22624–22638.
[7] H. Shi, Z. An, P.-Z. Li, J. Yin, G. Xing, T. He, H. Chen, J. Wang, H. Sun, W. Huang, Y. Zhao, Cryst. Growth Des. 2016, 16, 808–813.
[8] Q. Zhao, C. Huang, F. Li, Chem. Soc. Rev. 2011, 40, 2508–2524.
[9] M. K. Orloff, J. Chem. Phys. 1967, 47, 235–241.
[10] D. S. McClure, J. Chem. Phys. 1951, 19, 670–675.
[11] C. A. Parker, C. G. Hatchard, Trans. Faraday Soc. 1961, 57, 1894–1904.
[12] A. Maciejewski, M. Szymanski, R. P. Steer, J. Phys. Chem. 1986, 23, 6314–6318.
[13] B. Valeur, M. N. Berberan-Santos, Molecular Fluorescence 2012.
