
A2: Basics of photoelectron spectroscopy (PES)
Spectroscopy is one of the most important tools for gaining insights into the geometric and electronic structure of molecules and atoms. One method is photoelectron spectroscopy (PES), which provides information about the binding energies of electrons by inducing photoionization through excitation wavelengths of suitable energies. In contrast, the time resolved measurements provide an insight into the dynamic relaxation of the excited states. The information content depends crucially on the spectral resolution, which is why the development of femtosecond spectroscopy has contributed to the ability to investigate ultrafast molecular processes.[1,2] The dynamics of a molecule can be influenced by interactions with particles in the environment. For this reason, it makes sense to carry out experiments in the gas phase in order to investigate the intrinsic properties of the electronic structure.
External photoelectric effect & binding energy
The external photoelectric effect describes the phenomenon of electrons being detached from the surface of a metal by light irradiation. It was first observed in the 19th century and first interpreted by Albert Einstein in 1905.[3] He referred to Max Planck's quantum hypothesis on blackbody radiation (1900), according to which the energy of radiation is not emitted continuously, but in discrete energy packets, the so-called quanta.[4] The emitted energy Ε is therefore an integer multiple of an energy quantum, which is determined by the frequency ν of the radiation and Planck's quantum of action ℎ = 6.63·10-34 Js as a newly introduced natural constant: [4]
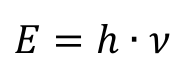
He also assumed that the energy of a light quantum that hits a metal surface is completely absorbed by an electron. Part of the energy is included in the work function Εw of the electron from the metal, while the rest appears as the kinetic energy Εkin of the ejected electron. This relationship is expressed by Einstein's equation: [3]
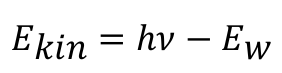
This behaviour can also be transferred to atoms and molecules. Instead of the work function required to release electrons from solids, in this case the binding energy ΕΒ must be overcome in order to detach electrons from the electron shell of the atom. From this context, Einstein's equation can be rewritten accordingly:
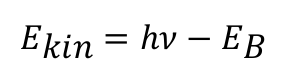
Depending on the initial charge of the particle, other terms are also used for the binding energy. The removal of an electron from a neutral or positively charged particle is referred to as ionisation energy. Similarly, electron affinity is used as a term for negatively charged particles.
By measuring the kinetic energy of the photoelectrons, the binding energies of the particles can be determined using Einstein’s equation. A theoretical estimate can be made using quantum mechanical calculations. For this purpose, Tjalling C. Koopmans developed a theory (Koopman’s theorem), which assumes in a first approximation that the electronic structure of a particle does not change during its photoionisation.[5] According to this theory, the binding energy corresponds to the energy of the orbital εi from which the electron was removed. The orbital energy εi results from the total energy difference between the initial state Εn and the final state Εn-1 of the system, which can be calculated using the Hartree-Fock method or density functional theory: [5]
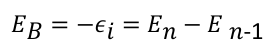
Franck-Condon principle & stationary PES
Due to photoionisation, the vibrational state of a molecule changes in addition to the electron state. For this purpose, the Franck-Condon principle is used to calculate the intensities of the possible transitions between the different vibrational states of different electronic states (see Figure 1). The principle is based on the Born-Oppenheimer approximation, so that the geometry of the core structure is assumed to be stationary, while the electronic transitions between the potential curves of the electronic states are vertical.

The vertical transition probability Ρ from the initial state i to the final state f is proportional to the square of the product of the transition dipole moment μfl→ and the electric field vector ε→ of the excitation light:
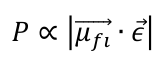
The transition dipole moment is calculated quantum mechanically using the equation:
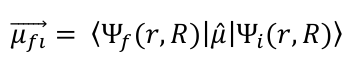
In applying the Born-Oppenheimer approximation, the wave function Ψ of the initial and final states can be separated into a nucleus-dependent (R) and an electron-dependent (r) part, which simplifies the integration over all coordinates. This is followed by the expression:
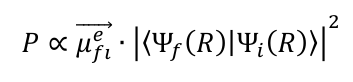
The equation above consists of two terms: the electronic transition dipole moment μefl→ and the subsequent Franck-Condon factor Ε = |〈Ψf (R)|Ψi (R)〉| 2 , which indicates the intensity with which a transition occurs after photoionisation. The greater the overlap integral of the vibrational wave functions, the more intense the transition. A resolution of the individual vibrational states is not possible with the femto-/nanosecond photoelectron spectrometer used in the working group of Prof. Dr. Kappes. The photoelectron spectrum therefore shows a continuous band whose maximum, however, can be assigned to the transition with the maximum overlap integral without knowing the exact final vibrational state reached. The associated binding energy is referred to as the vertical detachment energy (VDE). Another parameter in the photoelectron spectroscopic characterisation of molecules is the adiabatic detachment energy (ADE), which corresponds to the transition between the two vibrational ground states. It is the minimum energy required to remove an electron from the molecule and can be estimated by extrapolating the low-energy edge of the band.
Time-resolved PES
Dynamic relaxation processes from excited molecular states occur on ultrafast time scales and often involve structural and electronic changes. These can be investigated using time-resolved photoelectron spectroscopy in a pump-probe experiment. Here, the molecule is excited by the so-called pump pulse, and a weaker beam – referred to as the probe pulse – then monitors the response of the system. The photoelectron signal depends on the delay time between these two pulses, which can be varied in a targeted manner. The experimental resolution requires pulses with a temporal extent that is smaller than the temporal order of magnitude of the investigated processes. In the case of electronic states, pulse widths in the femtosecond range are required. Due to the energy-time uncertainty principle, temporal and energetic pulse widths are coupled with each other: [7]
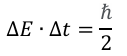
Accordingly, a compromise must be found between the temporal pulse width, which enables the time resolution of the processes, and the energetic pulse width. For this reason, pulse durations between 30 fs and 100 fs are useful for time-resolved processes in photoelectron spectroscopy.
From the excited state, various relaxation processes can take place, whereby intermolecular energy transfer processes are not significant in the gas phase due to the isolation of the molecules. The possible transitions are shown as a so-called Jablonski diagram (see Jablonski diagram).
References
[1] A. H. Zewail, J. Phys. Chem. A 2000, 104, 5660–5694.
[2] A. Stolow, A. E. Bragg, D. D. M Neumark, Chem. Rev. 2004, 104, 1719–1758.
[3] A. Einstein, Ann. Phys. 1905, 322, 132–148.
[4] M. Planck, Verh. Deutsch. Phys. Gesell. 1900, 2, 237–245.
[5] T. Koopmans, Physica 1934, 1, 104–113.
[6] V. Wiesiolek, Master Thesis, Karlsruher Institut für Technologie (KIT), 2023.
[7] W. Heisenberg, The physical principles of the quantum theory, Courier Corporation, 1949.
